Innate Immunity Cells and the Neurovascular Unit

 , ,
, , 
 , ,
, ,  and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
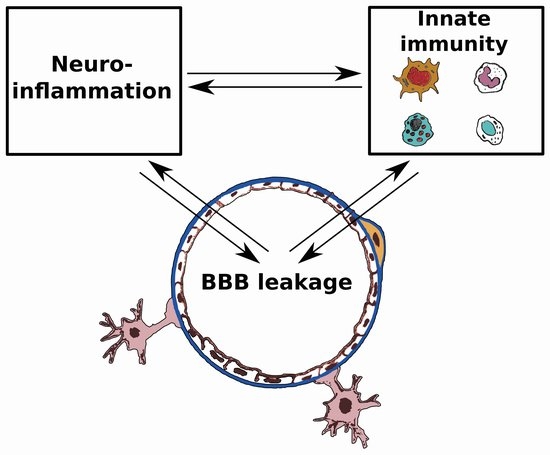
2. Basic Aspects of the Blood-Brain Barrier, Cells of Innate Immunity and Cerebral Edema
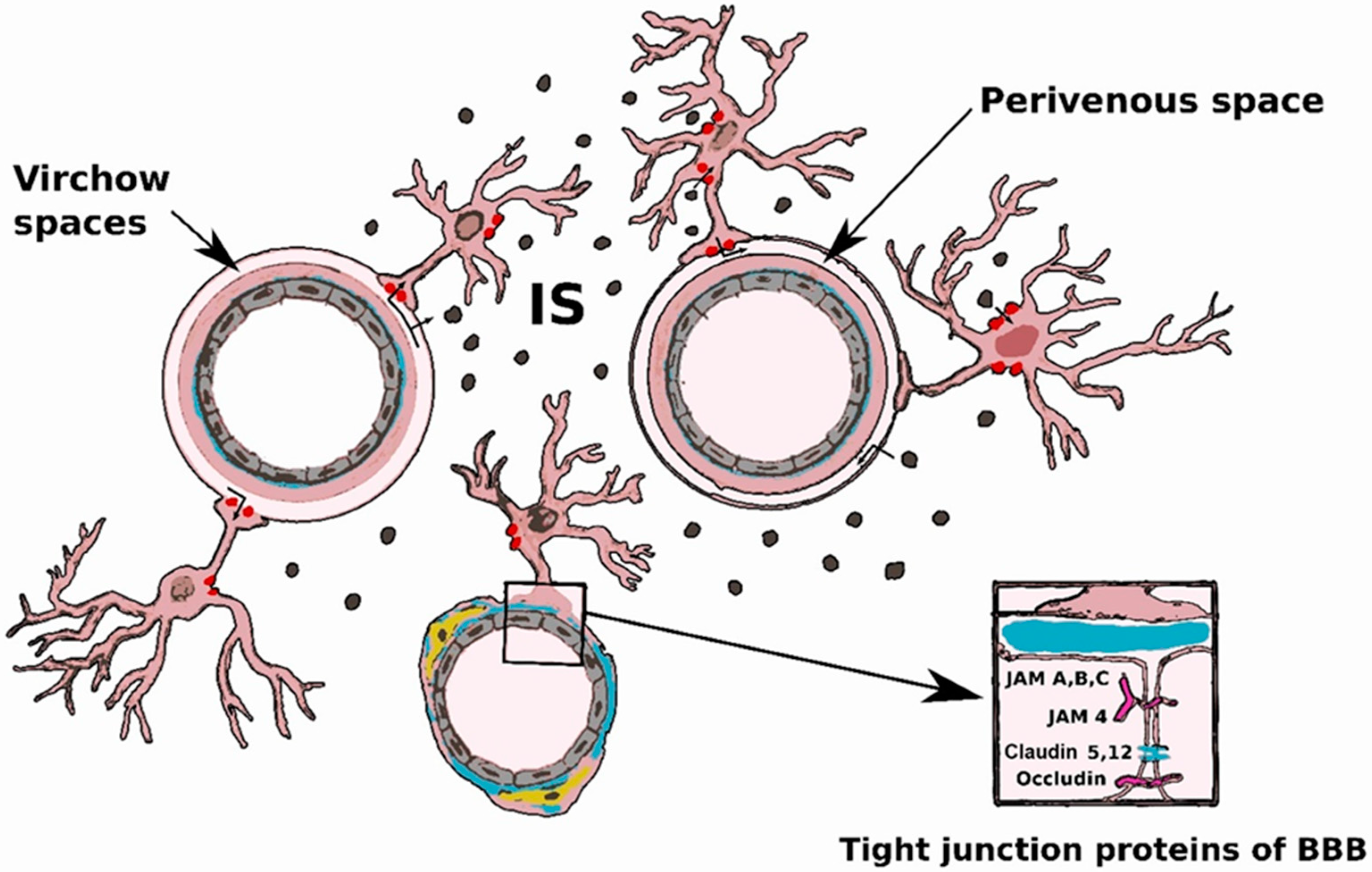
2.1. Blood-Brain Barrier
2.2. Innate Immunity Cells
2.3. Brain Edema
3. Blood-Brain Barrier (BBB) and Innate Immunity in Brain Pathology
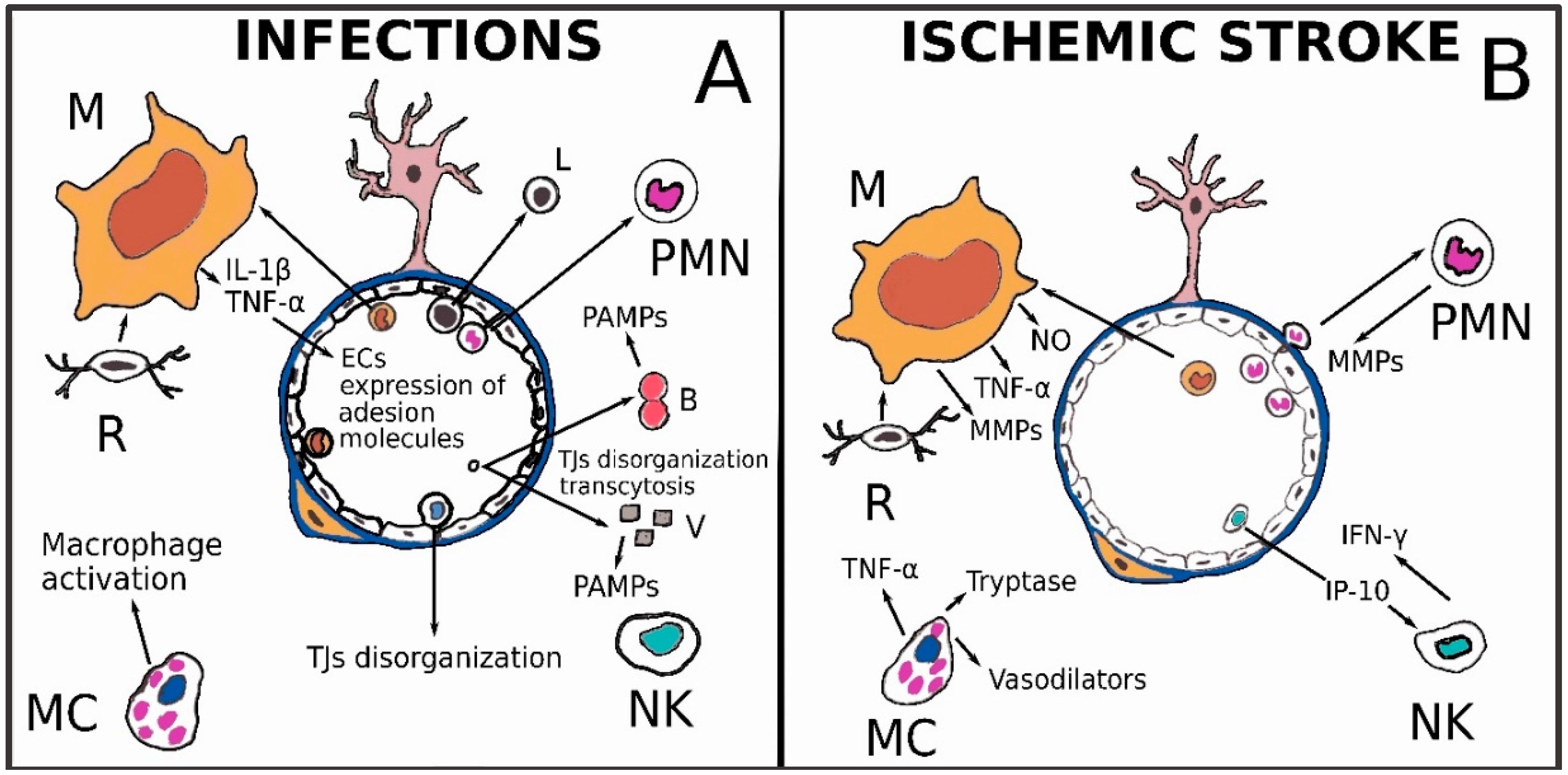
3.1. Infectious Diseases
3.2. Ischemic Stroke
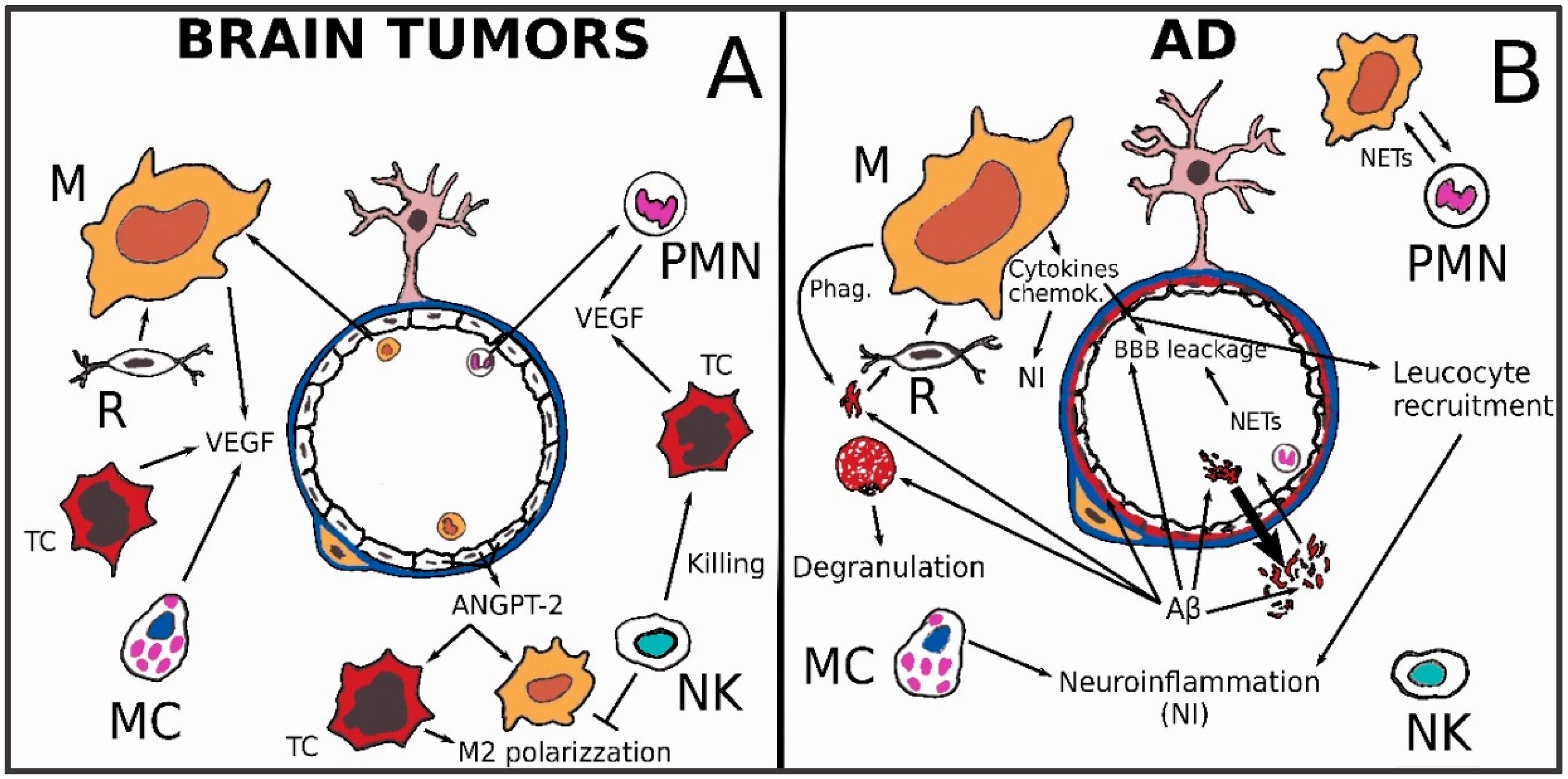
3.3. Brain Tumors
3.4. Alzheimer’s Disease
- early phase: excessive pro-inflammatory microglial activation that is maybe even precedent to AD’s clinical onset;
- late phase: substantial microglial impairment.
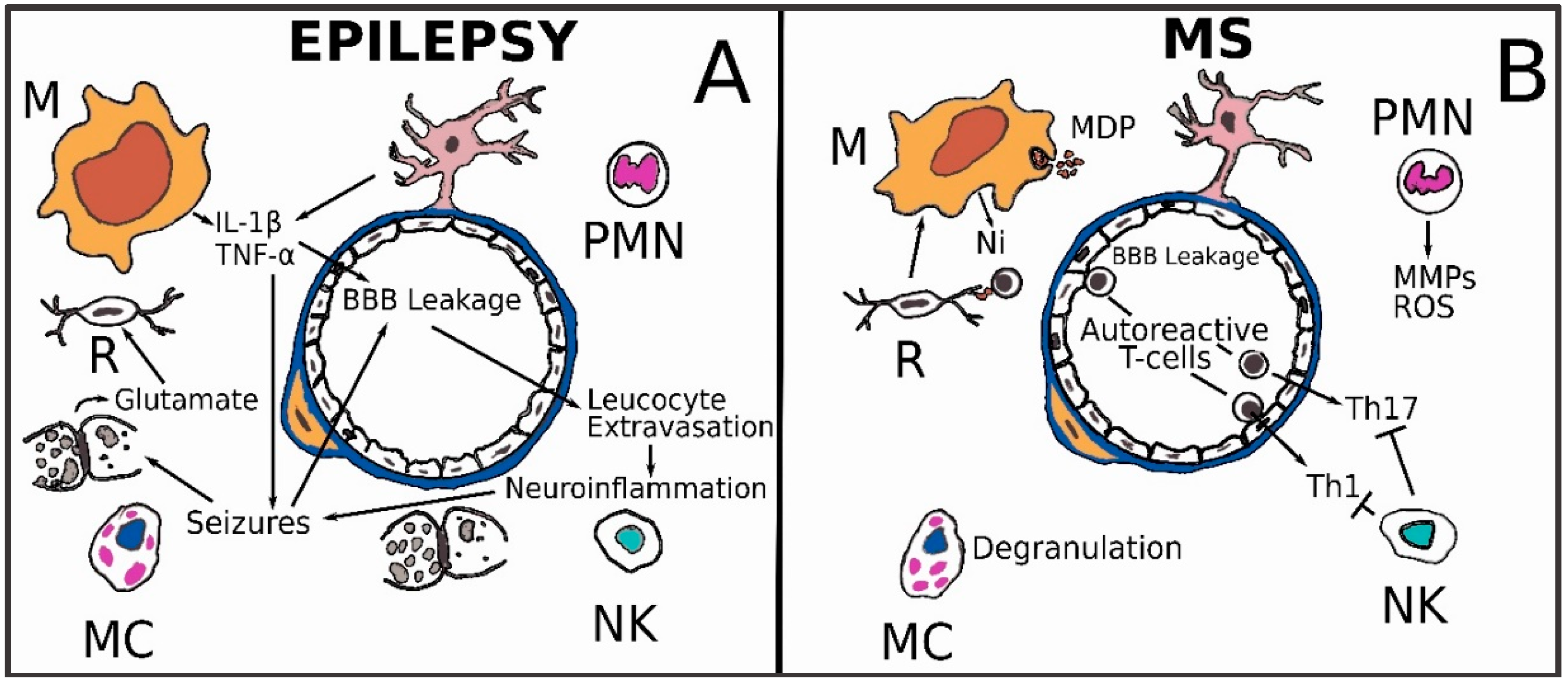
3.5. Epilepsy
3.6. Multiple Sclerosis
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BBB | Blood-brain barrier |
| BM | Basement membrane |
| BVES | Blood vessel epicardial substance |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| DAMPs | Damage-associated molecular patterns |
| EAE | Experimental autoimmune encephalomyelitis |
| ECM | Extracellular matrix |
| GDNF | Glial-derived neurotrophic factor |
| JAMs | Junctional adhesion molecule |
| MMPs | Metalloproteinases |
| NVU | Neuro-vascular unit |
| PAMPs | Pathogen-associated molecular patterns |
| pMCAO | Permanent middle cerebral artery occlusion |
| PMNs | Polymorphonuclear Neutrophils |
| PRRs | Pattern-recognition receptors |
| SMC | Smooth muscle cell |
| TAMs | Tumor associated macrophages |
| TMP | Trans-membrane protein |
References
- Fuller, G.N.; Burger, P.C. Central Nervous System. In Histology for Pathologists Lippincott; Mills, S.E., Ed.; Williams & Wilkins: Philadelphia, PA, USA, 2007; p. 305. ISBN 978-0-7817-6241-0. [Google Scholar]
- Rehfeld, A.; Nylander, M.; Karnov, K. Compendium of Histology; Springer: Cham, Switzerland, 2017; pp. 298–299. ISBN 978-3-319-41871-1. [Google Scholar]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef]
- Plog, B.A.; Nedergaard, M. The glymphatic system in central nervous system health and disease: Past, present, and future. Annu. Rev. Pathol. 2018, 13, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Verheggen, I.C.M.; Van Boxtel, M.P.J.; Verhey, F.R.J.; Jansen, J.F.A.; Backes, W.H. Interaction between blood-brain barrier and glymphatic system in solute clearance. Neurosci. Biobehav. Rev. 2018, 90, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Raab, S.; Beck, H.; Gaumann, A.; Yüce, A.; Gerber, H.P.; Plate, K.; Hammes, H.P.; Ferrara, N.; Breier, G. Impaired brain angiogenesis and neuronal apoptosis induced by conditional homozygous inactivation of vascular endothelial growth factor. Thromb. Haemost. 2004, 91, 595–605. [Google Scholar] [CrossRef]
- Schulzke, J.D.; Günzel, D.; John, L.J.; Fromm, M. Perspectives on tight junction research. Ann. N. Y. Acad. Sci. 2012, 1257, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H.; Kim, H.S.; Park, M.S.; Lee, E.B.; Lee, J.K.; Kim, J.T.; Kim, J.H.; Lee, M.C.; Lee, H.J.; Cho, K.H. Overexpression of caveolin-1 attenuates brain edema by inhibiting tight junction degradation. Oncotarget 2016, 7, 67857–67867. [Google Scholar] [CrossRef] [PubMed]
- Munde, P.B.; Khandekar, S.P.; Dive, A.M.; Upadhyaya, N.R. Pericytes in Health and Disease. Int. J. Oral Maxillofac. Pathol. 2014, 5, 2–7. [Google Scholar]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef]
- Cheslow, L.; Alvarez, J.I. Glial-endothelial crosstalk regulates blood-brain barrier function. Curr. Opin. Pharmacol. 2016, 26, 39–46. [Google Scholar] [CrossRef]
- Koehler, R.C.; Roman, R.J.; Harder, D.R. Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 2009, 32, 160–169. [Google Scholar] [CrossRef]
- Takeshita, Y.; Ransohoff, R.M. Inflammatory cell trafficking across the blood-brain barrier: Chemokine regulation and in vitro models. Immunol. Rev. 2012, 248, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Guadagno, E.; Presta, I.; Maisano, D.; Donato, A.; Pirrone, C.K.; Cardillo, G.; Corrado, S.D.; Mignogna, C.; Mancuso, T.; Donato, G.; et al. Role of macrophages in Brain Tumor Growth and Progression. Int. J. Mol. Sci. 2018, 19, 1005. [Google Scholar] [CrossRef]
- Perrotta, I.; Carito, V.; Russo, E.; Tripepi, S.; Aquila, S.; Donato, G. Macrophage autophagy and oxidative stress: An ultrastructural and immunoelectron microscopical study. Oxid. Med. Cell. Longev. 2011, 2011, 282739. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, I.; Brunelli, E.; Sciangula, A.; Conforti, F.; Perrotta, E.; Tripepi, S.; Donato, G.; Cassese, M. iNOS induction and PARP-1 activation in human atherosclerotic lesions: An immunohistochemical and ultrastructural approach. Cardiovasc. Pathol. 2011, 20, 195–203. [Google Scholar] [CrossRef]
- Di Vito, A.; Santise, G.; Mignogna, C.; Chiefari, E.; Cardillo, G.; Presta, I.; Arturi, F.; Malara, N.; Brunetti, F.; Donato, A.; et al. Innate immunity in cardiac myxomas and its pathological and clinical correlations. Innate Immun. 2018, 24, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.; Curley, J.P. Mast cells on the mind: New insights and opportunities. Trends Neurosci. 2013, 36, 513–521. [Google Scholar] [CrossRef]
- Mak, T.W.; Saunders, M.E.; Jett, B.D. Primer to the Immune Response, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 247–248. ISBN 9780123852458. [Google Scholar]
- Mak, T.W.; Saunders, M.E.; Jett, B.D. Primer to the Immune Response, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 24–25. ISBN 9780123852458. [Google Scholar]
- Nag, S.; Manias, J.L.; Stewart, D.J. Pathology and new players in the pathogenesis of brain edema. Acta Neuropathol. 2009, 118, 197–217. [Google Scholar] [CrossRef]
- Michinaga, S.; Koyama, Y. Pathogenesis of brain edema and investigation into anti-edema drugs. Int. J. Mol. Sci. 2015, 16, 9949–9975. [Google Scholar] [CrossRef]
- Brown, E.; Gray, F. Bacterial infections. In Greenfield’s Neuropathology, 8th ed.; Love, S., Louis, D.N., Ellison, D.W., Eds.; Arnold: London, UK, 2008; Volume 2, p. 1394. ISBN 9780340906811. [Google Scholar]
- Love, S.; Wiley, C.A. Viral Infections. In Greenfield’s Neuropathology, 8th ed.; Love, S., Louis, D.N., Ellison, D.W., Eds.; Arnold: London, UK, 2008; Volume 2, p. 1394. ISBN 9780340906811. [Google Scholar]
- Lampron, A.; Elali, A.; Rivest, S. Innate immunity in the CNS: Redefining the relationship between the CNS and Its environment. Neuron 2013, 8, 214–232. [Google Scholar] [CrossRef]
- Nakamuta, S.; Endo, H.; Higashi, Y.; Kousaka, A.; Yamada, H.; Yano, M.; Kido, H. Human immunodeficiency virus type 1 gp120-mediated disruption of tight junction proteins by induction of proteasome-mediated degradation of zonula occludens-1 and -2 in human brain microvascular endothelial cells. J. Neurovirol. 2008, 14, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Coureuil, M.; Mikaty, G.; Miller, F.; Lécuyer, H.; Bernard, C.; Bourdoulous, S.; Duménil, G.; Mège, R.M.; Weksler, B.B.; Romero, I.A.; et al. Meningococcal type IV pili recruit the polarity complex to cross the brain endothelium. Science 2009, 325, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Broadwell, R.D. Blood to brain and brain to blood passage of native horseradish peroxidase, wheat germ agglutinin, and albumin: Pharmacokinetic and morphological assessments. J. Neurochem. 1994, 62, 2404–2419. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.B.; Kim, K.S.; Bergelson, J.M. Poliovirus entry into human brain microvascular cells requires receptor-induced activation of SHP-2. EMBO J. 2007, 26, 4016–4028. [Google Scholar] [CrossRef] [PubMed]
- Roe, K.; Kumar, M.; Lum, S.; Orillo, B.; Nerurkar, V.R.; Verma, S. West Nile virus-induced disruption of the blood-brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases. J. Gen. Virol. 2012, 93, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Keaney, J.; Campbell, M. The dynamic blood-brain barrier. FEBS J. 2015, 282, 4067–4079. [Google Scholar] [CrossRef]
- Soulet, D.; Rivest, S. Microglia. Curr. Biol. 2008, 18, R506–R508. [Google Scholar] [CrossRef]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A.S. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin-4 and brain edema. Pediatr. Nephrol. 2007, 6, 778–784. [Google Scholar] [CrossRef]
- Sato, S.; Umenishi, F.; Inamasu, G.; Sato, M.; Ishikawa, M.; Nishizawa, M.; Oizumi, T. Expression of water channel mRNA following cerebral ischemia. Acta Neurochir. Suppl. 2000, 76, 239–241. [Google Scholar]
- Taniguchi, M.; Yamashita, T.; Kumura, E.; Tamatani, M.; Kobayashi, A.; Yokawa, T.; Maruno, M.; Kato, A.; Ohnishi, T.; Kohmura, E. Induction of aquaporin-4 water channel mRNA after focal cerebral ischemia in rat. Brain Res. Mol. Brain Res. 2000, 78, 131–137. [Google Scholar] [CrossRef]
- Gliem, M.; Krammes, K.; Liaw, L.; van Rooijen, N.; Hartung, H.P.; Jander, S. Macrophage-derived osteopontin induces reactive astrocyte polarization and promotes re-establishment of the blood brain barrier after ischemic stroke. Glia 2015, 63, 2198–2207. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhao, T.Z.; Zou, Y.J.; Zhang, J.H.; Feng, H. Hypoxia Induces autophagic cell death through hypoxia-inducible factor 1α in microglia. PLoS ONE 2014, 9, e96509. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Bonaventura, A.; Liberale, L.; Vecchié, A.; Casula, M.; Carbone, F.; Dallegri, F.; Montecucco, F. Update on Inflammatory Biomarkers and Treatments in Ischemic Stroke. Int. J. Mol. Sci. 2016, 17, 1967. [Google Scholar] [CrossRef]
- Breckwoldt, M.O.; Chen, J.W.; Stangenberg, L.; Aikawa, E.; Rodriguez, E.; Qiu, S.; Moskowitz, M.A.; Weissleder, R. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc. Nat. Acad. Sci. USA 2008, 105, 18584–18589. [Google Scholar] [CrossRef]
- Chu, H.X.; Broughton, B.R.; Kim, H.A.; Lee, S.; Drummond, G.R.; Sobey, C.G. Evidence that Ly6Chi monocytes are protective in acute ischemic stroke by promoting M2 macrophage polarization. Stroke 2015, 46, 1929–1937. [Google Scholar] [CrossRef]
- Zrzavy, T.; Machado-Santos, J.; Christine, S.; Baumgartner, C.; Weiner, H.L.; Butovsky, O.; Lassmann, H. Dominant role of microglial and macrophage innate immune responses in human ischemic infarcts. Brain Pathol. 2017. [Google Scholar] [CrossRef]
- Garcia, J.H.; Liu, K.F.; Bree, M.P. Effects of CD11b/18 monoclonal antibody on rats with permanent middle cerebral artery occlusion. Am. J. Pathol. 1996, 148, 241–248. [Google Scholar] [PubMed]
- Lopes Pinheiro, M.A.; Kooij, G.; Mizee, M.R.; Kamermans, A.; Enzmann, G.; Lyck, R.; Schwaninger, M.; Engelhardt, B.; de Vries, H.E. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim. Biophys. 2016, 1862, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Villa, P.; Triulzi, S.; Cavalieri, B.; Di Bitondo, R.; Bertini, R.; Barbera, S.; Bigini, P.; Mennini, T.; Gelosa, P.; Tremoli, E.; et al. The interleukin-8 (IL-8/CXCL8) receptor inhibitor reparixin improves neurological deficits and reduces long-term inflammation in permanent and transient cerebral ischemia in rats. Mol. Med. 2007, 13, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Perez-de-Puig, I.; Miró-Mur, F.; Ferrer-Ferrer, M.; Gelpi, E.; Pedragosa, J.; Justicia, C.; Urra, X.; Chamorro, A.; Planas, A.M. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. 2015, 129, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.J.; Sharp, F.R. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front. Cell. Neurosci. 2016, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Lindsberg, P.J.; Strbian, D.; Karjalainen-Lindsberg, M.L. Mast cells as early responders in the regulation of acute blood-brain barrier changes after cerebral ischemia and hemorrhage. J. Cereb. Blood Flow Metab. 2010, 30, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. The crucial role of mast cells in blood-brain barrier alterations. Exp. Cell. Res. 2015, 338, 119–125. [Google Scholar] [CrossRef]
- Peterfalvi, A.; Molnar, T.; Banati, M.; Pusch, G.; Miko, E.; Bogar, L.; Pal, J.; Szereday, L.; Illes, Z. Impaired function of innate T lymphocytes and NK cells in the acute phase of ischemic stroke. Cerebrovasc. Dis. 2009, 28, 490–498. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, Z.; Wang, D.; Zhang, T.; Sun, B.; Mu, L.; Wang, J.; Liu, Y.; Kong, Q.; Liu, X.; et al. Accumulation of natural killer cells in ischemic brain tissues and the chemotactic effect of IP-10. J. Neuroinflamm. 2014, 11, 79. [Google Scholar] [CrossRef]
- Wu, C.X.; Lin, G.S.; Lin, Z.X.; Zhang, J.D.; Liu, S.Y.; Zhou, C.F. Peritumoral edema shown by MRI predicts poor clinical outcome in glioblastoma. World J. Surg. Oncol. 2015, 13, 97. [Google Scholar] [CrossRef]
- Kerschbaumer, J.; Bauer, M.; Popovscaia, M.; Grams, A.E.; Thomé, C.; Freyschlag, C.F. Correlation of Tumor and Peritumoral Edema Volumes with Survival in Patients with Cerebral Metastases. Anticancer Res. 2017, 37, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Bastin, M.E.; Wardlaw, J.M.; Armitage, P.A.; Whittle, I.R. Effects of dexamethasone on peritumoral oedematous brain: A DT-MRI study. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1632–1635. [Google Scholar] [CrossRef] [PubMed]
- Kaal, E.C.; Vecht, C.J. The management of brain edema in brain tumors. Curr. Opin. Oncol. 2004, 6, 593–600. [Google Scholar] [CrossRef]
- Brandenburg, S.; Müller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 6, 977–982. [Google Scholar] [CrossRef]
- Turkowski, K.; Brandenburg, S.; Mueller, A.; Kremenetskaia, I.; Bungert, A.D.; Blank, A.; Felsenstein, M.; Vajkoczy, P. VEGF as a modulator of the innate immune response in glioblastoma. Glia 2018, 66, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Rozniecki, J.J.; Sahagian, G.; Jocobson, S.; Kempuraj, D.; Conti, P.; Kalogeromitros, D. Impact of stress and mast cells on brain metastases. J. Neuroimmunol. 2008, 205, 1–7. [Google Scholar] [CrossRef]
- Donato, G.; Conforti, F.; Camastra, C.; Ammendola, M.; Donato, A.; Renzulli, A. The role of mast cell tryptases in cardiac myxoma: Histogenesis and development of a challenging tumor. Oncol. Lett. 2014, 8, 379–383. [Google Scholar] [CrossRef]
- Markovic, M.; Antunovic, V.; Milenkovic, S.; Zivkovic, N. Prognostic value of peritumoral edema and angiogenesis in intracranial meningioma surgery. J. BUON 2013, 18, 430–436. [Google Scholar]
- Osawa, T.; Tosaka, M.; Nagaishi, M.; Yoshimoto, Y. Factors affecting peritumoral brain edema in meningioma: Special histological subtypes with prominently extensive edema. J. Neurooncol. 2013, 111, 49–57. [Google Scholar] [CrossRef]
- Donato, G.; Ferraro, G.; Signorelli, F.; Iofrida, G.; Lavano, A.; Amorosi, A.; Maltese, L.; Perrotta, I.; Tripepi, S.; Pardatscher, K.; et al. Chordoid meningioma: Case report and literature review. Ultrastruct. Pathol. 2006, 30, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Kshettry, V.R.; Selman, W.R.; Bambakidis, N.C. Peritumoral brain edema in intracranial meningiomas: The emergence of vascular endothelial growth factor-directed therapy. Neurosurg. Focus 2013, 35, E2. [Google Scholar] [CrossRef] [PubMed]
- Reszec, J.; Hermanowicz, A.; Rutkowski, R.; Bernaczyk, P.; Mariak, Z.; Chyczewski, L. Evaluation of mast cells and hypoxia inducible factor-1 expression in meningiomas of various grades in correlation with peritumoral brain edema. J. Neurooncol. 2013, 115, 119–125. [Google Scholar] [CrossRef]
- Polyzoidis, S.; Koletsa, T.; Panagiotidou, S.; Ashkan, K.; Theoharides, T.C. Mast cells in meningiomas and brain inflammation. J. Neuroinflamm. 2015, 12, 170. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S. Biology of natural killer cells in cancer and infection. Cancer Investig. 2002, 20, 405–419. [Google Scholar] [CrossRef]
- Krneta, T.; Gillgrass, A.; Poznanski, S.; Chew, M.; Lee, A.J.; Kolb, M.; Ashkar, A.A. M2-polarized and tumor-associated macrophages alter NK cell phenotype and function in a contact-dependent manner. J. Leukoc. Biol. 2017, 101, 285–295. [Google Scholar] [CrossRef]
- Castriconi, R.; Daga, A.; Dondero, A.; Zona, G.; Poliani, P.L.; Melotti, A.; Griffero, F.; Marubbi, D.; Spaziante, R.; Bellora, F.; et al. NK cells recognize and kill human glioblastoma cells with stem celllike properties. J. Immunol. 2009, 182, 3530–3539. [Google Scholar] [CrossRef]
- Avril, T.; Vauleon, E.; Hamlat, A.; Saikali, S.; Etcheverry, A.; Delmas, C.; Diabira, S.; Mosser, J.; Quillien, V. Human glioblastoma stem-like cells are more sensitive to allogeneic NK and T cell-mediated killing compared with serumcultured glioblastoma cells. Brain Pathol. 2012, 22, 159–174. [Google Scholar] [CrossRef]
- Heusinkveld, M.; van der Burg, S.H. Identification and manipulation of tumor associated macrophages in human cancers. J. Transl. Med. 2011, 9, 216. [Google Scholar] [CrossRef]
- Donato, G.; Iofrida, G.; Lavano, A.; Volpentesta, G.; Signorelli, F.; Pallante, P.L.; Berlingieri, M.T.; Pierantoni, M.G.; Palmieri, D.; Conforti, F.; et al. Analysis of UbcH10 expression represents a useful tool for the diagnosis and therapy of astrocytic tumors. Clin. Neuropathol. 2008, 27, 219–223. [Google Scholar] [CrossRef]
- Wang, W.; Cho, H.Y.; Rosenstein-Sisson, R.; Marín Ramos, N.I.; Price, R.; Hurth, K.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C. Intratumoral delivery of bortezomib: Impact on survival in an intracranial glioma tumor model. J. Neurosurg. 2018, 128, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Vengoji, R.; Macha, M.A.; Batra, S.K.; Shonka, N.A. Natural products: A hope for glioblastoma patients. Oncotarget 2018, 9, 22194–22219. [Google Scholar] [CrossRef] [PubMed]
- Bertaut, A.; Truntzer, C.; Madkouri, R.; Kaderbhai, C.G.; Derangère, V.; Vincent, J.; Chauffert, B.; Aubriot-Lorton, M.H.; Farah, W.; Mourier, K.L.; et al. Blood baseline neutrophil count predicts bevacizumab efficacy in glioblastoma. Oncotarget 2016, 7, 70948–70958. [Google Scholar] [CrossRef]
- Massara, M.; Persico, P.; Bonavita, O.; Mollica Poeta, V.; Locati, M.; Simonelli, M.; Bonecchi, R. Neutrophils in Gliomas. Front. Immunol. 2017, 8, 1349. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.W.; Choi, J.H.; Ahn, M.S.; Choi, Y.W.; Lee, H.W. Baseline neutrophil-lymphocyte ratio is associated with baseline and subsequent presence of brain metastases in advanced non-small-cell lung cancer. Sci. Rep. 2016, 6, 38585. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, I.; Nyúl-Tóth, Á.; Kozma, M.; Farkas, A.E.; Krizbai, I.A. Role of pattern recognition receptors of the neurovascular unit in inflamm-aging. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1000–H1012. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Clark, I.A.; Vissel, B. The meteorology of cytokine storms, and the clinical usefulness of this knowledge. Semin. Immunopathol. 2017, 39, 505–516. [Google Scholar] [CrossRef]
- De Felice, F.G.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef]
- Pickett, E.K.; Koffie, R.M.; Wegmann, S.; Henstridge, C.M.; Herrmann, A.G.; Colom-Cadena, M.; Lleo, A.; Kay, K.R.; Vaught, M.; Soberman, R.; et al. Non-fibrillar oligomeric amyloid-β within synapses. J. Alzheimer. Dis. 2016, 53, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Fadel, J.R.; Reagan, L.-P. Stop signs in hippocampal insulin signaling: The role of insulin resistance in structural, functional and behavioral deficits. Curr. Opin. Behav. Sci. 2016, 9, 47–54. [Google Scholar] [CrossRef]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Walter, F.S.; Kuhl, S.; Landmann, R.; Ishii, K.; Bertsch, T.; Stalder, A.K.; Muehlauser, F.; Liu, Y.; Ulmer, A.J.; et al. The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J. 2004, 18, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Koenigsknecht, J.; Landreth, G. Microglial phagocytosis of fibrillar β-amyloid through a β1 integrin-dependent mechanism. J. Neurosci. 2004, 24, 9838–9846. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef]
- Lee, C.Y.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef]
- Prokop, S.; Mille, K.R.; Heppner, F.L. Microglia actions in Alzheimer’s disease. Acta Neuropathol. 2013, 126, 461–477. [Google Scholar] [CrossRef]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef]
- Molteni, M.; Rossetti, C. Neurodegenerative diseases: The immunological perspective. J. Neuroimmunol. 2017, 313, 109–111. [Google Scholar] [CrossRef]
- Henry, R.J.; Kerr, D.M.; Finn, D.P.; Roche, M. For whom the endocannabinoid tolls: Modulation of innate immune function and implications for psychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Breitner, J.C.; Baker, L.D.; Montine, T.J.; Meinert, C.L.; Lyketsos, C.G.; Ashe, K.H.; Brandt, J.; Craft, S.; Evans, D.E.; Green, R.C.; et al. Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimer. Dement. 2011, 7, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Schafer, K.A.; Grundman, M.; Pfeiffer, E.; Sano, M.; Davis, K.L.; Farlow, M.R.; Jin, S.; Thomas, R.G.; Thal, L.J. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: A randomized controlled trial. JAMA 2003, 289, 2819–2826. [Google Scholar] [CrossRef] [PubMed]
- Leoutsakos, J.M.S.; Muthen, B.O.; Breitner, J.C.S.; Lyketsos, C.G. Effects of nonsteroidal anti-inflammatory drug treatments on cognitive decline vary by phase of pre-clinical Alzheimer disease: Findings from the randomized controlled Alzheimer’s disease anti-inflammatory prevention trial. Int. J. Geriatr. Psychiatry 2011, 27, 364–374. [Google Scholar] [CrossRef]
- Soininen, H.; West, C.; Robbins, J.; Niculescu, L. Long-term efficacy and safety of celecoxib in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2003, 23, 8–21. [Google Scholar] [CrossRef]
- Thal, L.J.; Ferris, S.H.; Kirby, L.; Block, G.A.; Lines, C.R.; Yuen, E.; Assaid, C.; Nessly, M.L.; Norman, B.A.; Baranak, C.C.; et al. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsycopharmacology 2005, 30, 1204–1215. [Google Scholar] [CrossRef]
- Da Fonseca, A.C.; Matias, D.; Garcia, C.; Amaral, R.; Geraldo, L.H.; Freitas, C.; Lima, F.R. The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 2014, 8, 362. [Google Scholar] [CrossRef]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef]
- Kumar, D.K.; Choi, S.H.; Washicosky, K.J.; Elmer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef]
- Van de Haar, H.J.; Burgmans, S.; Jansen, J.F.; Osch, M.J.; van Buchem, M.A.; Muller, M.; Hofman, P.A.; Verhey, F.R.; Backes, W.H. Blood-brain barrier leakage in patients with early Alzheimer’s disease. Radiology 2016, 152244. [Google Scholar] [CrossRef]
- Dose, J.; Huebbe, P.; Nebel, A.; Rimbach, G. APOE genotype and stress response—A mini review. Lipids Health Dis. 2016, 15, 121. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Cerebrovascular effects of apolipoprotein E: Implications for Alzheimer’s disease. JAMA Neurol. 2013, 70, 440–444. [Google Scholar] [CrossRef] [PubMed]
- VanItallie, T.B. Alzheimer’s disease: Innate immunity gone awry? Metabolism 2017, 69S, S41–S49. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2012, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Robinson, S.M.; Verma, S.; Morley, J.E. Efflux of human and mouse amyloid β proteins 1–40 and 1–42 from brain: Impairment in a mouse model of Alzheimer’s disease. Neuroscience 2003, 121, 487–492. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, M.A. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513. [Google Scholar] [CrossRef]
- Ashraf, T.; Kis, O.; Banerjee, N.; Bendayan, R. Drug transporters at brain barriers: Expression and regulation by neurological disorders. Adv. Exp. Med. Biol. 2012, 763, 20–69. [Google Scholar] [PubMed]
- Ravizza, T.; Vezzani, A. Status epilepticus induces time-dependent neuronal and astrocytic expression of interleukin-1 receptor type I in the rat limbic system. Neuroscience 2006, 137, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Boer, K.; van Vliet, E.A.; Redeker, S.; Baayen, J.C.; Spliet, W.G.; van Rijen, P.C.; Troost, D.; da Silva, F.H.; Wadman, W.J.; et al. Complement activation in experimental and human temporal lobe epilepsy. Neurobiology 2006, 26, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Ravizza, T.; Gagliardi, B.; Noé, F.; Boer, K.; Aronica, E.; Vezzani, A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 2008, 29, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Iori, V.; Frigerio, F.; Vezzani, A. Modulation of neuronal excitability by immune mediators in epilepsy. Curr. Opin. Pharmacol. 2016, 26, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Viviani, B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 2015, 96, 70–82. [Google Scholar] [CrossRef]
- Marchi, N.; Granata, T.; Janigro, D. Inflammatory pathways of seizure disorders. Trends Neurosci. 2014, 37, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Marchi, N.; Angelov, L.; Masaryk, T.; Fazio, V.; Granata, T.; Hernandez, N.; Hallene, K.; Diglaw, T.; Franic, L.; Najm, I.; et al. Seizure-promoting effect of blood-brain barrier disruption. Epilepsia 2007, 48, 732–742. [Google Scholar] [CrossRef]
- Gorter, J.A.; van Vliet, E.A.; Aronica, E. Status epilepticus, blood-brain barrier disruption, inflammation, and epileptogenesis. Epilepsy Behav. 2015, 49, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Kang, X.; Qiu, J.; Du, Y.; Jiang, J. Anti-Inflammatory Small Molecules to Treat Seizures and Epilepsy: From Bench to Bedside. Trends Pharmacol. Sci. 2016, 37, 463–484. [Google Scholar] [CrossRef]
- Fazio, F.; Ulivieri, M.; Volpi, C.; Gargaro, M.; Fallarino, F. Targeting metabotropic glutamate receptors for the treatment of neuroinflammation. Curr. Opin. Pharmacol. 2018, 38, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Scicchitano, F.; Citraro, R.; Aiello, R.; Camastra, C.; Mainardi, P.; Chimirri, S.; Perucca, E.; Donato, G.; De Sarro, G. Protective activity of α-lactoalbumin (ALAC), a whey protein rich in tryptophan, in rodent models of epileptogenesis. Neuroscience 2012, 13, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, E.A.; Otte, W.M.; Wadman, W.J.; Aronica, E.; Kooij, G.; de Vries, H.E.; Dijkhuizen, R.M.; Gorter, J.A. Blood-brain barrier leakage after status epilepticus in rapamycin-treated rats I: Magnetic resonance imaging. Epilepsia 2016, 57, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Citraro, R.; Donato, G.; Camastra, C.; Iuliano, R.; Cuzzocrea, S.; Constanti, A.; De Sarro, G. mTOR inhibition modulates epileptogenesis, seizures and depressive behavior in a genetic rat model of absence epilepsy. Neuropharmacology 2013, 69, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Follesa, P.; Citraro, R.; Camastra, C.; Donato, A.; Isola, D.; Constanti, A.; De Sarro, G.; Donato, G. The mTOR signaling pathway and neuronal stem/progenitor cell proliferation in the hippocampus are altered during the development of absence epilepsy in a genetic animal model. Neurol. Sci. 2014, 35, 1793–1799. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Pedro, N.Y.; Espinosa-Ramirez, G.; de la Cruz, V.P.; Pineda, B.; Sotelo, J. Initial immunopathogenesis of multiple sclerosis: Innate immune response. Clin. Dev. Immunol. 2013, 2013, 413465. [Google Scholar] [CrossRef] [PubMed]
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Medical progress: Multiple sclerosis—The plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955. [Google Scholar] [CrossRef]
- Nylander, A.; Haer, D.A. Multiple sclerosis. J. Clin. Investig. 2012, 122, 1180–1188. [Google Scholar] [CrossRef]
- Babbe, H.; Roers, A.; Waisman, A.; Lassmann, H.; Goebels, N.; Hohlfeld, R.; Friese, M.; Schröder, R.; Deckert, M.; Schmidt, S.; et al. Clonal expansions of CD8(+) T cells dominate the T cell infi ltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 2000, 192, 393–404. [Google Scholar] [CrossRef]
- Jacobsen, M.; Cepok, S.; Quak, E.; Happel, M.; Gaber, R.; Ziegler, A.; Schock, S.; Oertel, W.H.; Sommer, N.; Hemmer, B. Oligoclonal expansion of memory CD8+ T cells in cerebrospinal fluid from multiple sclerosis patients. Brain 2002, 125, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.; Laroni, A.; Weiner, H.L. Role of the innate immune system in the pathogenesis of multiple sclerosis. J. Neuroimmunol. 2010, 221, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, B.; Kerschensteiner, M.; Korn, T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015, 144, 406–419. [Google Scholar] [CrossRef]
- Eitan, S.; Zisling, R.; Cohen, A.; Belkin, M.; Hirschberg, D.L.; Lotan, M.; Schwartz, M. Identification of an interleukin 2-like substance as a factor cytotoxic to oligodendrocytes and associated with central nervous system regeneration. Proc. Natl. Acad. Sci. USA 1992, 89, 5442–5446. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.P.; Barnett, M.H.; Parratt, J.D.; Prineas, J.W. Multiple sclerosis: Distribution of inflammatory cells in newly forming lesions. Ann. Neurol. 2009, 66, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Thomas, T.L.; Betmouni, S.; Scolding, N.; Love, S. Elevated activity and microglial expression of myeloperoxidase in demyelinated cerebral cortex in multiple sclerosis. Brain Pathol. 2008, 18, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Raivich, G.; Banati, R. Brain microglia and blood-derived macrophages: Molecular profiles and functional roles in multiple sclerosis and animal models of autoimmune demyelinating disease. Brain Res. Brain Res. Rev. 2004, 46, 261–281. [Google Scholar] [CrossRef]
- Sørensen, T.L.; Tani, M.; Jensen, J.; Pierce, V.; Lucchinetti, C.; Folcik, V.A.; Qin, S.; Rottman, J.; Sellebjerg, F.; Strieter, R.M.; et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J. Clin. Investig. 1999, 103, 807–815. [Google Scholar] [CrossRef]
- Aravalli, R.N.; Peterson, P.K.; Lokensgard, J.R. Toll-like receptors in defense and damage of the central nervous system. J. Neuroimmune Pharmacol. 2007, 2, 297–312. [Google Scholar] [CrossRef]
- Jack, C.S.; Arbour, N.; Blain, M.; Meier, U.C.; Prat, A.; Antel, J.P. Th1 polarization of CD4+ T cells by Toll-like receptor 3-activated human microglia. J. Neuropathol. Exp. Neurol. 2007, 66, 848–859. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, S. Toll-like receptors and inflammation in the CNS. Curr. Drug Targets Inflamm. Allergy 2002, 1, 181–191. [Google Scholar] [PubMed]
- Andersson, A.; Covacu, R.; Sunnemark, D.; Danilov, A.I.; Dal Bianco, A.; Khademi, M.; Wallstrom, E.; Lobell, A.; Brundin, L.; Lassmann, H.; et al. Pivotal advance: HMGB1 expression in active lesions of human and experimental multiple sclerosis. J. Leukoc. Biol. 2008, 84, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Bsibsi, M.; Ravid, R.; Gveric, D.; van Noort, J.M. Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Sloane, J.A.; Batt, C.; Ma, Y.; Harris, Z.M.; Trapp, B.; Vartanian, T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc. Natl. Acad. Sci. USA 2010, 107, 11555–11560. [Google Scholar] [CrossRef] [PubMed]
- Crowley, T.; Fitzpatrick, J.M.; Kuijper, T.; Cryan, J.F.; O’Toole, O.; O’Leary, O.F.; Downer, E.J. Modulation of TLR3/TLR4 inflammatory signaling by the GABAB receptor agonist baclofen in glia and immune cells: Relevance to therapeutic effects in multiple sclerosis. Front. Cell. Neurosci. 2015, 9, 284. [Google Scholar] [CrossRef] [PubMed]
- Olsson, Y. Mast cells in plaques of multiple sclerosis. Acta Neurol. Scand. 1974, 50, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Toms, R.; Weiner, H.L.; Johnson, D. Identification of IgE-positive cells and mast cells in frozen sections of multiple sclerosis brains. J. Neuroimmunol. 1990, 30, 169–177. [Google Scholar] [CrossRef]
- Rozniecki, J.J.; Hauser, S.L.; Stein, M.; Lincoln, R.; Theoharides, T.C. Elevated mast cell tryptase in cerebrospinal fluid of multiple sclerosis patients. Ann. Neurol. 1995, 37, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Lock, C.; Hermans, G.; Pedotti, R.; Brendolan, A.; Schadt, E.; Garren, H.; Langer-Gould, A.; Strober, S.; Cannella, B.; Allard, J.; et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 2002, 8, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Weinberg, R.B. Mast Cells and innate Lymphoid Cells: Underappreciated Players in CNS Autoimmune Demyelinating Disease. Front. Immunl. 2018, 9, 514. [Google Scholar] [CrossRef]
- Russi, A.E.; Walker-Cauleld, M.E.; Guo, Y.; Lucchinetti, C.F.; Brown, M.A. Meningeal mast cell-T cell crosstalk regulates T cell encephalitogenicity. J. Autoimmun. 2016, 73, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Codarri, L.; Gyulveszi, G.; Tosevski, V.; Hesske, L.; Fontana, A.; Magnenat, L.; Suter, T.; Becher, B. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the e ector phase of autoimmune neuroin ammation. Nat. Immunol. 2011, 12, 560–567. [Google Scholar] [CrossRef] [PubMed]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.; Cullimore, M.; Safavi, F.; Zhang, G.X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 2011, 12, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, E.D.; Shriver, L.P.; Maresz, K.; Pedras-Vasconcelos, J.; Verthelyi, D.; Dittel, B.N. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J. Immunol. 2007, 178, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Croxford, A.L.; Lanzinger, M.; Hartmann, F.J.; Schreiner, B.; Mair, F.; Pelczar, P.; Clausen, B.E.; Jung, S.; Greter, M.; Becher, B. The cytokine GM-CSF drives the in ammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity 2015, 43, 502–514. [Google Scholar] [CrossRef]
- Lefrançais, E.; Duval, A.; Mirey, E.; Roga, S.; Espinosa, E.; Cayrol, C.; Girard, J.P. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2014, 111, 15502–15507. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.T.; Martin, M.U. Interleukin 33 is a guardian of barriers and a local alarmin. Nat. Immunol. 2016, 17, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Russi, A.E.; Ebel, M.E.; Yang, Y.; Brown, M.A. Male-specific IL-33 expression regulates sex-dimorphic EAE susceptibility. Proc. Natl. Acad. Sci. USA 2018, 115, E1520–E1529. [Google Scholar] [CrossRef]
- Logothetis, L.; Mylonas, I.A.; Baloyannis, S.; Pashalidou, M.; Orologas, A.; Zafeiropoulos, A.; Kosta, V.; Theoharides, T.C. A pilot, open label, clinical trial using hydroxyzine in multiple sclerosis. Int. J. Immunopathol. Pharmacol. 2005, 18, 771–778. [Google Scholar] [CrossRef]
- Kempuraj, D.; Tagen, M.; Iliopoulou, B.P.; Clemons, A.; Vasiadi, M.; Boucher, W.; House, M.; Wolfberg, A.; Theoharides, T.C. Luteolin inhibits myelin basic protein-induced human mast cell activation and mast cell-dependent stimulation of Jurkat T cells. Br. J. Pharmacol. 2008, 155, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Kempuraj, D.; Kourelis, T.; Manola, A. Human mast cells stimulate activated T cells: Implications for multiple sclerosis. Ann. N. Y. Acad. Sci. 2008, 1144, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, T.L.; Liu, R.; Price, M.; Rhodes, S.; La Cava, A.; Shi, F.D. Differential effects of IL-21 during initiation and progression of autoimmunity against neuroantigen. J. Immunol. 2005, 174, 2696–2701. [Google Scholar] [CrossRef] [PubMed]
- Winkler-Pickett, R.; Young, H.A.; Cherry, J.M.; Diehl, J.; Wine, J.; Back, T.; Bere, W.E.; Mason, A.T.; Ortaldo, J.R. In vivo regulation of experimental autoimmune encephalomyelitis by NK cells: Alteration of primary adaptive responses. J Immunol. 2008, 180, 4495–4506. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Fazekas, G.; Hara, H.; Tabira, T. Mechanism of natural killer (NK) cell regulatory role in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 163, 24–30. [Google Scholar] [CrossRef]
- Galazka, G.; Jurewicz, A.; Orlowski, W.; Stasiolek, M.; Brosnan, C.F.; Raine, C.S.; Selmaj, K. EAE tolerance induction with Hsp70-peptide complexes depends on H60 and NKG2D activity. J. Immunol. 2007, 179, 4503–4515. [Google Scholar] [CrossRef]
- Huang, D.; Shi, F.D.; Jung, S.; Pien, G.C.; Wang, J.; Salazar-Mather, T.P.; He, T.T.; Weaver, J.T.; Ljunggren, H.G.; Biron, C.A.; et al. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 2006, 20, 896–905. [Google Scholar] [CrossRef]
- Traugott, U. Characterization and distribution of lymphocyte subpopulations in multiple sclerosis plaques versus autoimmune demyelinating lesions. Springer Semin. Immunopathol. 1985, 8, 71–95. [Google Scholar] [CrossRef]
- Kastrukoff, L.F.; Lau, A.; Wee, R.; Zecchini, D.; White, R.; Paty, D.W. Clinical relapses of multiple sclerosis are associated with ‘novel’ valleys in natural killer cell functional activity. J. Neuroimmunol. 2003, 145, 103–114. [Google Scholar] [CrossRef]
- Kastrukoff, L.F.; Morgan, N.G.; Zecchini, D.; White, R.; Petkau, A.J.; Satoh, J.; Paty, D.W. A role for natural killer cells in the immunopathogenesis of multiple sclerosis. J. Neuroimmunol. 1998, 86, 123–133. [Google Scholar] [CrossRef]
- Takahashi, K.; Miyake, S.; Kondo, T.; Terao, K.; Hatakenaka, M.; Hashimoto, S.; Yamamura, T. Natural killer type 2 bias in remission of multiple sclerosis. J. Clin. Investig. 2001, 107, R23–R29. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Aranami, T.; Endoh, M.; Miyake, S.; Yamamura, T. The regulatory role of natural killer cells in multiple sclerosis. Brain 2004, 127, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.; Siracusa, M.C.; Yap, G.S.; Gause, W.C. Innate cell communication kick-starts pathogen-specific immunity. Nat. Immunol. 2016, 17, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Moynihan, K.D.; Irvine, D.J. Roles for Innate Immunity in Combination Immunotherapies. Cancer Res. 2017, 77, 5215–5221. [Google Scholar] [CrossRef] [PubMed]
- Faissner, S.; Gold, R. Correction to: Efficacy and Safety of the Newer Multiple Sclerosis Drugs Approved Since 2010. CNS Drugs. 2018, 32, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Prcina, M.; Novak, M.; Cigankova, V.; Kontsekova, E. Immunosenescence—The role in the immunotherapy of older population. Bratisl. Lek. Listy 2018, 119, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, E.M. Tau Immunotherapy. Neurodegener. Dis. 2016, 16, 34–38. [Google Scholar] [CrossRef]
- Iwasaki, A. Immune Regulation of Antibody Access to Neuronal Tissues. Trends Mol. Med. 2017, 23, 227–245. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Presta, I.; Vismara, M.F.M.; Novellino, F.; Donato, A.; Zaffino, P.; Scali, E.; Pirrone, K.C.; Spadea, M.F.; Malara, N.; Donato, G. Innate Immunity Cells and the Neurovascular Unit. Int. J. Mol. Sci. 2018, 19, 3856. https://doi.org/10.3390/ijms19123856
Presta I, Vismara MFM, Novellino F, Donato A, Zaffino P, Scali E, Pirrone KC, Spadea MF, Malara N, Donato G. Innate Immunity Cells and the Neurovascular Unit. International Journal of Molecular Sciences. 2018; 19(12):3856. https://doi.org/10.3390/ijms19123856
Chicago/Turabian StylePresta, Ivan, Marco Flavio Michele Vismara, Fabiana Novellino, Annalidia Donato, Paolo Zaffino, Elisabetta Scali, Krizia Caterina Pirrone, Maria Francesca Spadea, Natalia Malara, and Giuseppe Donato. 2018. "Innate Immunity Cells and the Neurovascular Unit" International Journal of Molecular Sciences 19, no. 12: 3856. https://doi.org/10.3390/ijms19123856
APA StylePresta, I., Vismara, M. F. M., Novellino, F., Donato, A., Zaffino, P., Scali, E., Pirrone, K. C., Spadea, M. F., Malara, N., & Donato, G. (2018). Innate Immunity Cells and the Neurovascular Unit. International Journal of Molecular Sciences, 19(12), 3856. https://doi.org/10.3390/ijms19123856